Ácido desoxirribonucleico
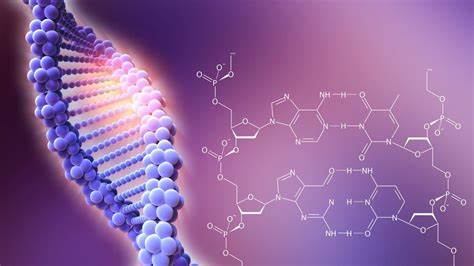
El ácido desoxirribonucleico, conocido también por las siglas ADN, es un ácido nucleico que contiene las instrucciones genéticas usadas en el desarrollo y funcionamiento de todos los organismos vivos y algunos virus; también es responsable de la transmisión hereditaria. La función principal de la molécula de ADN es el almacenamiento a largo plazo de información para construir otros componentes de las células, como las proteínas y las moléculas de ARN. Los segmentos de ADN que llevan esta información genética son llamados genes, pero las otras secuencias de ADN tienen propósitos estructurales o toman parte en la regulación del uso de esta información genética.
Desde el punto de vista químico, el ADN es un polímero de nucleótidos, es decir, un polinucleótido. Cada nucleótido, a su tiempo, está formado por un glúcido (la desoxirribosa), una base nitrogenada (que puede ser adenina→A, timina→T, citosina→C o guanina→G) y un grupo fosfato (derivado del ácido fosfórico). Lo que distingue a un polinucleótido de otro es, entonces, la base nitrogenada, y por ello la secuencia del ADN se especifica nombrando solo la secuencia de sus bases. La disposición secuencial de estas cuatro bases a lo largo de la cadena es la que codifica la información genética, siguiendo el siguiente criterio de complementariedad: A-T y G-C. Esto se debe a que la adenina y la guanina son de mayor tamaño que la timina y la citosina, por lo que este criterio permite cumplir una uniformidad. En los organismos vivos, el ADN se presenta como una doble cadena de nucleótidos, en la que las dos hebras están unidas entre sí por unas conexiones denominadas puentes de hidrógeno.
Para que la información que contiene el ADN pueda ser utilizada por la maquinaria celular debe copiarse en primer lugar en unos trenes de nucleótidos, más cortos y con unas unidades diferentes, llamados ARN. Las moléculas de ARN se copian exactamente del ADN mediante un proceso denominado transcripción. Una vez procesadas en el núcleo celular, las moléculas de ARN pueden salir al citoplasma para su utilización posterior. La información contenida en el ARN se interpreta usando el código genético, que especifica la secuencia de los aminoácidos de las proteínas, según una correspondencia de un triplete de nucleótidos (codón) para cada aminoácido. Esto es, la información genética (esencialmente: qué proteínas se van a producir en cada momento del ciclo de vida de una célula) se halla codificada en las secuencias de nucleótidos del ADN y debe traducirse para poder funcionar. Tal traducción se realiza usando el código genético a modo de diccionario. El diccionario "secuencia de nucleótido-secuencia de aminoácidos" permite el ensamblado de largas cadenas de aminoácidos (las proteínas) en el citoplasma de la célula. Por ejemplo, en el caso de la secuencia de ADN indicada antes (ATGCTAGCATCG...), la ADN polimerasa utilizaría como molde la cadena complementaria de dicha secuencia de ADN (que sería TAC-GAT-CGT-AGC-...) para transcribir una molécula de ARNm que se leería AUG-CUA-GCA-UCG-...; el ARNm resultante, utilizando el código genético, se traduciría como la secuencia de aminoácidos metionina-leucina-ácido aspártico-arginina-...
Las secuencias de ADN que constituyen la unidad fundamental, física y funcional de la herencia se denominan genes. Cada gen contiene una parte que se transcribe a ARN y otra que se encarga de definir cuándo y dónde deben expresarse. La información contenida en los genes (genética) se emplea para generar ARN y proteínas, que son los componentes básicos de las células, los "ladrillos" que se utilizan para la construcción de los orgánulos u organelos celulares, entre otras funciones.
Dentro de las células, el ADN está organizado en estructuras llamadas cromosomas que, durante el ciclo celular, se duplican antes de que la célula se divida. Los organismos eucariotas (por ejemplo, animales, plantas y hongos) almacenan la mayor parte de su ADN dentro del núcleo celular y una mínima parte en elementos celulares llamados mitocondrias, y en los plastos y los centros organizadores de microtúbulos o centríolos, en caso de tenerlos; los organismos procariotas (bacterias y arqueas) lo almacenan en el citoplasma de la célula y, por último, los virus ADN lo hacen en el interior de la cápside de naturaleza proteica. Existen multitud de proteínas, como por ejemplo las histonas y los factores de transcripción, que se unen al ADN dotándolo de una estructura tridimensional determinada y regulando su expresión. Los factores de transcripción reconocen secuencias reguladoras del ADN y especifican la pauta de transcripción de los genes. El material genético completo de una dotación cromosómica se denomina genoma y, con pequeñas variaciones, es característico de cada especie.

El ADN fue aislado por primera vez durante 1869, por el médico suizo Friedrich Miescher, mientras trabajaba en la Universidad de Tubinga. Miescher realizaba experimentos acerca de la composición química del pus de vendas quirúrgicas desechadas cuando notó un precipitado de una sustancia desconocida que caracterizó químicamente más tarde. Lo llamó nucleína, debido a que lo había extraído a partir de núcleos celulares. Se necesitaron casi 70 años de investigación para poder identificar los componentes y la estructura de los ácidos nucleicos.
En 1919 Phoebus Levene identificó que un nucleótido está formado por una base nitrogenada, un azúcar y un fosfato. Levene sugirió que el ADN generaba una estructura con forma de solenoide (muelle) con unidades de nucleótidos unidos a través de los grupos fosfato. En 1930 Levene y su maestro Albrecht Kossel probaron que la nucleína de Miescher es un ácido desoxirribonucleico (ADN) formado por cuatro bases nitrogenadas (citosina (C), timina (T), adenina (A) y guanina (G), el azúcar desoxirribosa y un grupo fosfato, y que, en su estructura básica, el nucleótido está compuesto por un azúcar unido a la base y al fosfato. Sin embargo, Levene pensaba que la cadena era corta y que las bases se repetían en un orden fijo. En 1937 William Astbury produjo el primer patrón de difracción de rayos X que mostraba que el ADN tenía una estructura regular.
La función biológica del ADN comenzó a dilucidarse en 1928, con una serie básica de experimentos de la genética moderna realizados por Frederick Griffith, quien estaba trabajando con cepas «lisas» (S) o «rugosas» (R) de la bacteria Pneumococcus (causante de la neumonía), según la presencia (S) o no (R) de una cápsula azucarada, que es la que confiere virulencia (véase también experimento de Griffith). La inyección de neumococos S vivos en ratones produce la muerte de estos, y Griffith observó que, si inyectaba ratones con neumococos R vivos o con neumococos S muertos por calor, los ratones no morían. Sin embargo, si inyectaba a la vez neumococos R vivos y neumococos S muertos, los ratones morían, y en su sangre se podían aislar neumococos S vivos. Como las bacterias muertas no pudieron haberse multiplicado dentro del ratón, Griffith razonó que debía producirse algún tipo de cambio o transformación de un tipo bacteriano a otro por medio de una transferencia de alguna sustancia activa, que denominó principio transformante. Esta sustancia proporcionaba la capacidad a los neumococos R de producir una cápsula azucarada y transformarse así en virulentas. En los siguientes 15 años, estos experimentos iniciales se replicaron mezclando distintos tipos de cepas bacterianas muertas por el calor con otras vivas, tanto en ratones (in vivo) como en tubos de ensayo (in vitro). La búsqueda del «factor transformante» que era capaz de hacer virulentas a cepas que inicialmente no lo eran continuó hasta 1944, año en el cual Oswald Avery, Colin MacLeod y Maclyn McCarty realizaron un experimento hoy clásico. Estos investigadores extrajeron la fracción activa (el factor transformante) y, mediante análisis químicos, enzimáticos y serológicos, observaron que no contenía proteínas, ni lípidos no ligados, ni polisacáridos activos, sino que estaba constituido principalmente por "una forma viscosa de ácido desoxirribonucleico altamente polimerizado", es decir, ADN. El ADN extraído de las cepas bacterianas S muertas por el calor lo mezclaron "in vitro" con cepas R vivas: el resultado fue que se formaron colonias bacterianas S, por lo que se concluyó inequívocamente que el factor o principio transformante era el ADN.
A pesar de que la identificación del ADN como principio transformante aún tardó varios años en ser universalmente aceptada, este descubrimiento fue decisivo en el conocimiento de la base molecular de la herencia, y constituye el nacimiento de la genética molecular. Finalmente, el papel exclusivo del ADN en la heredabilidad fue confirmado en 1952 mediante los experimentos de Alfred Hershey y Martha Chase, en los cuales comprobaron que el fago T2 transmitía su información genética en su ADN, pero no en su proteína (véase también experimento de Hershey y Chase).
En cuanto a la caracterización química de la molécula, Chargaff realizó en 1940 algunos experimentos que le sirvieron para establecer las proporciones de las bases nitrogenadas en el ADN. Descubrió que las proporciones de purinas eran idénticas a las de pirimidinas, la «equimolecularidad» de las bases ([A]=[T], [G]=[C]) y el hecho de que la cantidad de G+C en una determinada molécula de ADN no siempre es igual a la cantidad de A+T y puede variar desde el 36 hasta el 70 por ciento del contenido total. Con toda esta información y junto con los datos de difracción de rayos X proporcionados por Rosalind Franklin, James Watson y Francis Crick propusieron en 1953 el modelo de la doble hélice de ADN para representar la estructura tridimensional del polímero. En una serie de cinco artículos en el mismo número de Nature se publicó la evidencia experimental que apoyaba el modelo de Watson y Crick. De estos, el artículo de Franklin y Raymond Gosling fue la primera publicación con datos de difracción de rayos X que apoyaba el modelo de Watson y Crick, y en ese mismo número de Nature también aparecía un artículo sobre la estructura del ADN de Maurice Wilkins y sus colaboradores.
Watson, Crick y Wilkins recibieron conjuntamente, en 1962, después de la muerte de Rosalind Franklin, el Premio Nobel en Fisiología o Medicina. Sin embargo, el debate continúa sobre quién debería recibir crédito por el descubrimiento.

El ADN es un largo polímero formado por unidades repetitivas, los nucleótidos. Una doble cadena de ADN mide de 22 a 26 ángstroms (2,2 a 2,6 nanómetros) de ancho, y una unidad (un nucleótido) mide 3,3 Å (0,33 nm) de largo. Aunque cada unidad individual que se repite es muy pequeña, los polímeros de ADN pueden ser moléculas enormes que contienen millones de nucleótidos. Por ejemplo, el cromosoma humano más largo, el cromosoma número 1, tiene aproximadamente 220 millones de pares de bases.
En los organismos vivos, el ADN no suele existir como una molécula individual, sino como una pareja de moléculas estrechamente asociadas. Las dos cadenas de ADN se enroscan sobre sí mismas formando una especie de escalera de caracol, denominada doble hélice. El modelo de estructura en doble hélice fue propuesto en 1953 por James Watson y Francis Crick (el artículo «Molecular Structure of Nucleic Acids: A Structure for Deoxyribose Nucleic Acid» fue publicado el 25 de abril de 1953 en Nature), después de obtener una imagen de la estructura de doble hélice gracias a la refracción por rayos X hecha por Rosalind Franklin. El éxito de este modelo radicaba en su consistencia con las propiedades físicas y químicas del ADN. El estudio mostraba, además, que la complementariedad de bases podía ser relevante en su replicación, y también la importancia de la secuencia de bases como portadora de información genética. Cada unidad que se repite, el nucleótido, contiene un segmento de la estructura de soporte (azúcar + fosfato), que mantiene la cadena unida, y una base, que interacciona con la otra cadena de ADN en la hélice. En general, una base ligada a un azúcar se denomina nucleósido y una base ligada a un azúcar y a uno o más grupos fosfatos recibe el nombre de nucleótido.
Cuando muchos nucleótidos se encuentran unidos, como ocurre en el ADN, el polímero resultante se denomina polinucleótido.
Estructura de soporte: La estructura de soporte de una hebra de ADN está formada por unidades alternas de grupos fosfato y azúcar (desoxirribosa). El azúcar en el ADN es una pentosa, concretamente, la desoxirribosa.
También existen otras bases nitrogenadas (las llamadas bases nitrogenadas minoritarias), derivadas de forma natural o sintética de alguna otra base mayoritaria. Lo son por ejemplo la hipoxantina, relativamente abundante en el tRNA, o la cafeína, ambas derivadas de la adenina; otras, como el aciclovir, derivadas de la guanina, son análogos sintéticos usados en terapia antiviral; otras, como una de las derivadas del uracilo, son antitumorales.
Las bases nitrogenadas tienen una serie de características que les confieren unas propiedades determinadas. Una característica importante es su carácter aromático, consecuencia de la presencia en el anillo de dobles enlaces en posición conjugada. Ello les confiere la capacidad de absorber luz en la zona ultravioleta del espectro en torno a los 260 nm, lo cual puede aprovecharse para determinar el coeficiente de extinción del ADN y hallar la concentración existente de los ácidos nucleicos. Otra de sus características es que presentan tautomería o isomería de grupos funcionales, debido a que un átomo de hidrógeno unido a otro átomo puede migrar a una posición vecina; en las bases nitrogenadas se dan dos tipos de tautomerías: tautomería lactama-lactima, donde el hidrógeno migra del nitrógeno al oxígeno del grupo oxo (forma lactama) y viceversa (forma lactima), y tautomería imina-amina primaria, donde el hidrógeno puede estar formando el grupo amina (forma amina primaria) o migrar al nitrógeno adyacente (forma imina). La adenina solamente puede presentar tautomería amina-imina, la timina y el uracilo muestran tautomería doble lactama-lactima, y la guanina y citosina pueden presentar ambas. Por otro lado, y aunque se trate de moléculas apolares, las bases nitrogenadas presentan suficiente carácter polar como para establecer puentes de hidrógeno, ya que tienen átomos muy electronegativos (nitrógeno y oxígeno) que presentan carga parcial negativa, y átomos de hidrógeno con carga parcial positiva, de manera que se forman dipolos que permiten que se formen estos enlaces débiles.

Se estima que el genoma humano haploide tiene alrededor de 3000 millones de pares de bases. Para indicar el tamaño de las moléculas de ADN se indica el número de pares de bases, y como derivados hay dos unidades de medida muy utilizadas, la kilobase (kb), que equivale a 1000 pares de bases, y la megabase (Mb), que equivale a un millón de pares de bases.
La dóble hélice de ADN se mantiene estable mediante la formación de puentes de hidrógeno entre las bases asociadas a cada una de las dos hebras. Para la formación de un enlace de hidrógeno una de las bases debe presentar un "donador" de hidrógenos con un átomo de hidrógeno con carga parcial positiva (-NH2 o -NH) y la otra base debe presentar un grupo "aceptor" de hidrógenos con un átomo cargado electronegativamente (C=O o N). Los puentes de hidrógeno son uniones más débiles que los típicos enlaces químicos covalentes, como los que conectan los átomos en cada hebra de ADN, pero más fuertes que interacciones hidrófobas individuales, enlaces de Van der Waals, etc. Como los puentes de hidrógeno no son enlaces covalentes, pueden romperse y formarse de nuevo de forma relativamente sencilla. Por esta razón, las dos hebras de la doble hélice pueden separarse como una cremallera, bien por fuerza mecánica o por alta temperatura. La doble hélice se estabiliza además por el efecto hidrofóbico y el apilamiento, que no se ven influidos por la secuencia de bases del ADN.
Cada tipo de base en una hebra forma un enlace únicamente con un tipo de base en la otra hebra, lo que se denomina complementariedad de las bases. Así, las purinas forman enlaces con las pirimidinas, de forma que A se enlaza solo con T, y C solo con G. La organización de dos nucleótidos apareados a lo largo de la doble hélice se denomina apareamiento de bases. Este emparejamiento corresponde a la observación ya realizada por Erwin Chargaff (1905-2002), que mostró que la cantidad de adenina era muy similar a la cantidad de timina, y que la cantidad de citosina era igual a la cantidad de guanina en el ADN. Como resultado de esta complementariedad, toda la información contenida en la secuencia de doble hebra de la hélice de ADN está duplicada en cada hebra, lo cual es fundamental durante el proceso de replicación del ADN. En efecto, esta interacción reversible y específica entre pares de bases complementarias es crítica para todas las funciones del ADN en los organismos vivos.
Como se ha indicado anteriormente, los dos tipos de pares de bases forman un número diferente de enlaces de hidrógeno: A=T forman dos puentes de hidrógeno, y C≡G forman tres puentes de hidrógeno (ver imágenes). El par de bases GC es por tanto más fuerte que el par de bases AT. Como consecuencia, tanto el porcentaje de pares de bases GC como la longitud total de la doble hélice de ADN determinan la fuerza de la asociación entre las dos hebras de ADN. Las dobles hélices largas de ADN con alto contenido en GC tienen hebras que interaccionan más fuertemente que las dobles hélices cortas con alto contenido en AT. Por esta razón, las zonas de la doble hélice de ADN que necesitan separarse fácilmente tienden a tener un alto contenido en AT, como por ejemplo la secuencia TATAAT de la caja de Pribnow de algunos promotores. En el laboratorio, la fuerza de esta interacción puede medirse buscando la temperatura requerida para romper los puentes de hidrógeno, la temperatura de fusión (también denominado valor Tm, del inglés melting temperature). Cuando todas las pares de bases en una doble hélice se funden, las hebras se separan en solución en dos hebras completamente independientes. Estas moléculas de ADN de hebra simple no tienen una única forma común, sino que algunas conformaciones son más estables que otras.
Existen diferentes tipos de pares de bases que se pueden formar según el modo como se forman los puentes de hidrógeno. Los que se observan en la doble hélice de ADN son los llamados pares de bases Watson-Crick, pero también existen otros posibles pares de bases, como los denominados Hoogsteen y Wobble u oscilante, que pueden aparecer en circunstancias particulares. Además, para cada tipo existe a su vez el mismo par reverso, es decir, el que se da si se gira la base pirimidínica 180º sobre su eje.
En total, en su forma tautomérica mayoritaria, existen 28 posibles pares de bases nitrogenadas: 10 posibles pares de bases purina-pirimidina (2 pares Watson-Crick y 2 Watson Crick reverso, 1 par Hoogsteen y 2 pares Hoogsteen reverso, 1 par oscilante y 2 pares oscilante reverso), 7 pares homo purina-purina (A=A, G=G), 4 pares A=G y 7 pares pirimidina-pirimidina. Esto sin contar con los pares de bases que pueden formarse si también tenemos en cuenta las otras formas tautoméricas minoritarias de las bases nitrogenadas; estos, además, pueden ser responsables de mutaciones puntuales por sustitución de tipo transición.

El ADN es una molécula bicatenaria, es decir, está formada por dos cadenas dispuestas de forma antiparalela y con las bases nitrogenadas enfrentadas. En su estructura tridimensional, se distinguen distintos niveles:
El ADN existe en muchas conformaciones. Sin embargo, en organismos vivos sólo se han observado las conformaciones ADN-A, ADN-B y ADN-Z. La conformación que adopta el ADN depende de su secuencia, la cantidad y dirección de superenrollamiento que presenta, la presencia de modificaciones químicas en las bases y las condiciones de la solución, tales como la concentración de iones de metales y poliaminas. De las tres conformaciones, la forma "B" es la más común en las condiciones existentes en las células. Las dos dobles hélices alternativas del ADN difieren en su geometría y dimensiones.
La forma "A" es una espiral que gira hacia la derecha, más amplia que la "B", con una hendidura menor superficial y más amplia, y una hendidura mayor más estrecha y profunda. La forma "A" ocurre en condiciones no fisiológicas en formas deshidratadas de ADN, mientras que en la célula puede producirse en apareamientos híbridos de hebras ADN-ARN, además de en complejos enzima-ADN.
Los segmentos de ADN en los que las bases han sido modificadas por metilación pueden sufrir cambios conformacionales mayores y adoptar la forma "Z". En este caso, las hebras giran alrededor del eje de la hélice en una espiral que gira a mano izquierda, lo opuesto a la forma "B" más frecuente. Estas estructuras poco frecuentes pueden ser reconocidas por proteínas específicas que se unen a ADN-Z y posiblemente estén implicadas en la regulación de la transcripción.
En los extremos de los cromosomas lineales existen regiones especializadas de ADN denominadas telómeros. La función principal de estas regiones es permitir a la célula replicar los extremos cromosómicos utilizando la enzima telomerasa, puesto que las enzimas que replican el resto del ADN no pueden copiar los extremos 3' de los cromosomas. Estas terminaciones cromosómicas especializadas también protegen los extremos del ADN, y evitan que los sistemas de reparación del ADN en la célula los procesen como ADN dañado que debe ser corregido. En las células humanas, los telómeros son largas zonas de ADN de hebra sencilla que contienen algunos miles de repeticiones de una única secuencia TTAGGG.
Estas secuencias ricas en guanina pueden estabilizar los extremos cromosómicos mediante la formación de estructuras de juegos apilados de unidades de cuatro bases, en lugar de los pares de bases encontrados normalmente en otras estructuras de ADN. En este caso, cuatro bases guanina forman unidades con superficie plana que se apilan una sobre otra, para formar una estructura cuádruple-G estable. Estas estructuras se estabilizan formando puentes de hidrógeno entre los extremos de las bases y la quelatación de un metal iónico en el centro de cada unidad de cuatro bases. También se pueden formar otras estructuras, con el juego central de cuatro bases procedente, o bien de una hebra sencilla plegada alrededor de las bases, o bien de varias hebras paralelas diferentes, de forma que cada una contribuye con una base a la estructura central.

Además de estas estructuras apiladas, los telómeros también forman largas estructuras en lazo, denominadas lazos teloméricos o lazos-T (T-loops en inglés). En este caso, las hebras simples de ADN se enroscan sobre sí mismas en un amplio círculo estabilizado por proteínas que se unen a telómeros. En el extremo del lazo T, el ADN telomérico de hebra sencilla se sujeta a una región de ADN de doble hebra porque la hebra de ADN telomérico altera la doble hélice y se aparea a una de las dos hebras. Esta estructura de triple hebra se denomina lazo de desplazamiento o lazo D (D-loop).
La doble hélice es una espiral dextrógira, esto es, cada una de las cadenas de nucleótidos gira a la derecha; esto puede verificarse si nos fijamos, yendo de abajo arriba, en la dirección que siguen los segmentos de las hebras que quedan en primer plano. Si las dos hebras giran a derechas se dice que la doble hélice es dextrógira, y si giran a izquierdas, levógira (esta forma puede aparecer en hélices alternativas debido a cambios conformacionales en el ADN). Pero en la conformación más común que adopta el ADN, la doble hélice es dextrógira, girando cada par de bases respecto al anterior unos 36º.
Cuando las dos hebras de ADN se enrollan una sobre la otra (sea a derechas o a izquierdas), se forman huecos o hendiduras entre una hebra y la otra, dejando expuestos los laterales de las bases nitrogenadas del interior (ver la animación). En la conformación más común que adopta el ADN aparecen, como consecuencia de los ángulos formados entre los azúcares de ambas cadenas de cada par de bases nitrogenadas, dos tipos de hendiduras alrededor de la superficie de la doble hélice: una de ellas, la hendidura o surco mayor, que mide 22 Å (2,2 nm) de ancho, y la otra, la hendidura o surco menor, que mide 12 Å (1,2 nm) de ancho. Cada vuelta de hélice, que es cuando esta ha realizado un giro de 360º o lo que es lo mismo, de principio de hendidura mayor a final de hendidura menor, medirá por tanto 34 Å, y en cada una de esas vueltas hay unos 10,5 pb.
La anchura de la hendidura mayor implica que los extremos de las bases son más accesibles en esta, de forma que la cantidad de grupos químicos expuestos también es mayor lo cual facilita la diferenciación entre los pares de bases A-T, T-A, C-G, G-C. Como consecuencia de ello, también se verá facilitado el reconocimiento de secuencias de ADN por parte de diferentes proteínas sin la necesidad de abrir la doble hélice. Así, proteínas como los factores de transcripción que pueden unirse a secuencias específicas, frecuentemente contactan con los laterales de las bases expuestos en la hendidura mayor. Por el contrario, los grupos químicos que quedan expuestos en la hendidura menor son similares, de forma que el reconocimiento de los pares de bases es más difícil; por ello se dice que la hendidura mayor contiene más información que la hendidura menor.
Una secuencia de ADN se denomina «sentido» (en inglés, sense) si su secuencia es la misma que la secuencia de un ARN mensajero que se traduce en una proteína. La secuencia de la hebra de ADN complementaria se denomina «antisentido» (antisense). En ambas hebras de ADN de la doble hélice pueden existir tanto secuencias «sentido», que codifican ARNm, como «antisentido», que no lo codifican. Es decir, las secuencias que codifican ARNm no están todas presentes en una sola de las hebras, sino repartidas entre las dos hebras. Tanto en procariotas como en eucariotas se producen ARN con secuencias antisentido, pero la función de esos ARN no está completamente clara. Se ha propuesto que los ARN antisentido están implicados en la regulación de la expresión génica mediante apareamiento ARN-ARN: los ARN antisentido se aparearían con los ARNm complementarios, bloqueando de esta forma su traducción.
En unas pocas secuencias de ADN en procariotas y eucariotas —este hecho es más frecuente en plásmidos y virus—, la distinción entre hebras sentido y antisentido es más difusa, debido a que presentan genes superpuestos. En estos casos, algunas secuencias de ADN tienen una función doble, codificando una proteína cuando se lee a lo largo de una hebra, y una segunda proteína cuando se lee en la dirección contraria a lo largo de la otra hebra. En bacterias, esta superposición puede estar involucrada en la regulación de la transcripción del gen, mientras que en virus los genes superpuestos aumentan la cantidad de información que puede codificarse en sus diminutos genomas.
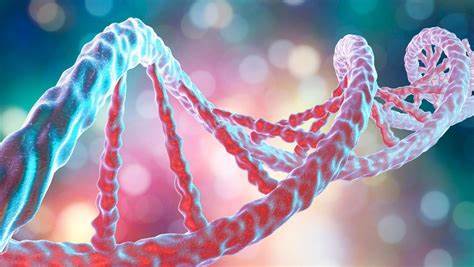
El ADN puede retorcerse como una cuerda en un proceso que se denomina superenrollamiento del ADN (supercoiling, en inglés). Cuando el ADN está en un estado "relajado", una hebra normalmente gira alrededor del eje de la doble hélice una vez cada 10,4 pares de bases, pero si el ADN está retorcido las hebras pueden estar unidas más estrechamente o más relajadamente. Si el ADN está retorcido en la dirección de la hélice, se dice que el superenrollamiento es positivo, y las bases se mantienen juntas de forma más estrecha. Si el ADN se retuerce en la dirección opuesta, el superenrollamiento se llama negativo, y las bases se alejan. En la naturaleza, la mayor parte del ADN tiene un ligero superenrollamiento negativo que es producido por enzimas denominadas topoisomerasas. Estas enzimas también son necesarias para liberar las fuerzas de torsión introducidas en las hebras de ADN durante procesos como la transcripción y la replicación.
La expresión de los genes está influenciada por la forma en la que el ADN está empaquetado en cromosomas, en una estructura denominada cromatina. Las modificaciones de bases pueden estar implicadas en el empaquetamiento del ADN: las regiones que presentan una expresión génica baja o nula normalmente contienen niveles altos de metilación de las bases citosina. Por ejemplo, la metilación de citosina produce 5-metil-citosina, que es importante para la inactivación del cromosoma X. El nivel medio de metilación varía entre organismos: el gusano Caenorhabditis elegans carece de metilación de citosina, mientras que los vertebrados presentan un nivel alto —hasta 1 %— de su ADN contiene 5-metil-citosina. A pesar de la importancia de la 5-metil-citosina, esta puede desaminarse para generar una base timina. Las citosinas metiladas son por tanto particularmente sensibles a mutaciones. Otras modificaciones de bases incluyen la metilación de adenina en bacterias y la glicosilación de uracilo para producir la "base-J" en kinetoplastos.
El ADN puede resultar dañado por muchos tipos de mutágenos, que cambian la secuencia del ADN: agentes alquilantes, además de radiación electromagnética de alta energía, como luz ultravioleta y rayos X. El tipo de daño producido en el ADN depende del tipo de mutágeno. Por ejemplo, la luz UV puede dañar al ADN produciendo dímeros de timina, que se forman por ligamiento cruzado entre bases pirimidínicas. Por otro lado, oxidantes tales como radicales libres o el peróxido de hidrógeno producen múltiples daños, incluyendo modificaciones de bases, sobre todo guanina, y roturas de doble hebra (double-strand breaks). En una célula humana cualquiera, alrededor de 500 bases sufren daño oxidativo cada día. De estas lesiones oxidativas, las más peligrosas son las roturas de doble hebra, ya que son difíciles de reparar y pueden producir mutaciones puntuales, inserciones y deleciones de la secuencia de ADN, así como translocaciones cromosómicas.
Muchos mutágenos se posicionan entre dos pares de bases adyacentes, por lo que se denominan agentes intercalantes. La mayoría de los agentes intercalantes son moléculas aromáticas y planas, como el bromuro de etidio, la daunomicina, la doxorubicina y la talidomida. Para que un agente intercalante pueda integrarse entre dos pares de bases, estas deben separarse, distorsionando las hebras de ADN y abriendo la doble hélice. Esto inhibe la transcripción y la replicación del ADN, causando toxicidad y mutaciones. Por ello, los agentes intercalantes del ADN son a menudo carcinógenos: el benzopireno, las acridinas, la aflatoxina y el bromuro de etidio son ejemplos bien conocidos. Sin embargo, debido a su capacidad para inhibir la replicación y la transcripción del ADN, estas toxinas también se utilizan en quimioterapia para inhibir el rápido crecimiento de las células cancerosas.
El daño en el ADN inicia una respuesta que activa diferentes mecanismos de reparación que reconocen lesiones específicas en el ADN, que son reparadas en el momento para recuperar la secuencia original del ADN. Asimismo, el daño en el ADN provoca una parada en el ciclo celular, que conlleva la alteración de numerosos procesos fisiológicos, que a su vez implica síntesis, transporte y degradación de proteínas (véase también Checkpoint de daños en el ADN). Alternativamente, si el daño genómico es demasiado grande para que pueda ser reparado, los mecanismos de control inducirán la activación de una serie de rutas celulares que culminarán en la muerte celular.
Las funciones biológicas del ADN incluyen el almacenamiento de información (genes y genoma), la codificación de proteínas (transcripción y traducción) y su autoduplicación (replicación del ADN) para asegurar la transmisión de la información a las células hijas durante la división celular.
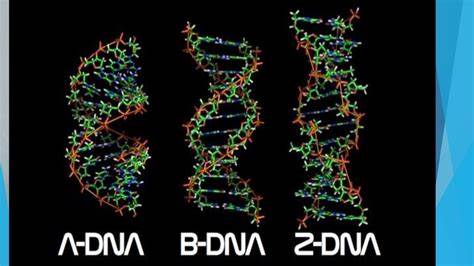
El ADN se puede considerar como un almacén cuyo contenido es la información (mensaje) necesaria para construir y sostener el organismo en el que reside, la cual se transmite de generación en generación. El conjunto de información que cumple esta función en un organismo dado se denomina genoma, y el ADN que lo constituye, ADN genómico.
El ADN genómico (que se organiza en moléculas de cromatina que a su vez se ensamblan en cromosomas) se encuentra en el núcleo celular de los eucariotas, además de pequeñas cantidades en las mitocondrias y cloroplastos. En procariotas, el ADN se encuentra en un cuerpo de forma irregular denominado nucleoide.
La información de un genoma está contenida en los genes, y al conjunto de toda la información que corresponde a un organismo se le denomina su genotipo. Un gen es una unidad de herencia y es una región de ADN que influye en una característica particular de un organismo (como el color de los ojos, por ejemplo). Los genes contienen un "marco de lectura abierto" (open reading frame) que puede transcribirse, además de secuencias reguladoras, tales como promotores y enhancers, que controlan la transcripción del marco de lectura abierto.
Desde este punto de vista, las obreras de este mecanismo son las proteínas. Estas pueden ser estructurales, como las proteínas de los músculos, cartílagos, pelo, etc., o funcionales, como la hemoglobina o las innumerables enzimas del organismo. La función principal de la herencia es la especificación de las proteínas, siendo el ADN una especie de plano o receta para producirlas. La mayor parte de las veces la modificación del ADN provocará una disfunción proteica que dará lugar a la aparición de alguna enfermedad. Pero en determinadas ocasiones, las modificaciones podrán provocar cambios beneficiosos que darán lugar a individuos mejor adaptados a su entorno.
Las aproximadamente treinta mil proteínas diferentes en el cuerpo humano están constituidas por veinte aminoácidos diferentes, y una molécula de ADN debe especificar la secuencia en que se unen dichos aminoácidos.
En el proceso de elaborar una proteína, el ADN de un gen se lee y se transcribe a ARN. Este ARN sirve como mensajero entre el ADN y la maquinaria que elaborará las proteínas y por eso recibe el nombre de ARN mensajero o ARNm. El ARN mensajero sirve de molde a la maquinaria que elabora las proteínas, para que ensamble los aminoácidos en el orden preciso para armar la proteína.

El dogma central de la biología molecular establecía que el flujo de actividad y de información era: ADN → ARN → proteína. No obstante, en la actualidad ha quedado demostrado que este "dogma" debe ser ampliado, pues se han encontrado otros flujos de información: en algunos organismos (virus de ARN) la información fluye de ARN a ADN; este proceso se conoce como "transcripción inversa o reversa", también llamada "retrotranscripción". Además, se sabe que existen secuencias de ADN que se transcriben a ARN y son funcionales como tales, sin llegar a traducirse nunca a proteína: son los ARN no codificantes, como es el caso de los ARN interferentes.
El ADN del genoma de un organismo puede dividirse conceptualmente en dos: el que codifica las proteínas (los genes) y el que no codifica. En muchas especies, solo una pequeña fracción del genoma codifica proteínas. Por ejemplo, solo alrededor del 1,5 % del genoma humano consiste en exones que codifican proteínas (20 000 a 25 000 genes), mientras que más del 90 % consiste en ADN no codificante.
El ADN no codificante (también denominado ADN basura o junk DNA) corresponde a secuencias del genoma que no generan una proteína (procedentes de transposiciones, duplicaciones, translocaciones y recombinaciones de virus, etc.), incluyendo los intrones. Hasta hace poco tiempo se pensaba que el ADN no codificante no tenía utilidad alguna, pero estudios recientes indican que eso es inexacto. Entre otras funciones, se postula que el llamado "ADN basura" regula la expresión diferencial de los genes. Por ejemplo, algunas secuencias tienen afinidad hacia proteínas especiales que tienen la capacidad de unirse al ADN (como los homeodominios, los complejos receptores de hormonas esteroides, etc.), con un papel importante en el control de los mecanismos de trascripción y replicación. Estas secuencias se llaman frecuentemente "secuencias reguladoras", y los investigadores suponen que solo se ha identificado una pequeña fracción de las que realmente existen. La presencia de tanto ADN no codificante en genomas eucarióticos y las diferencias en tamaño del genoma entre especies representan un misterio que es conocido como el "enigma del valor de C". Los elementos repetitivos también son elementos funcionales. Si no se considerasen así, se excluiría más del 50 % de los nucleótidos totales, ya que constituyen elementos de repetición. Recientemente, un grupo de investigadores de la Universidad de Yale ha descubierto una secuencia de ADN no codificante que sería la responsable de que los seres humanos hayan desarrollado la capacidad de agarrar y/o manipular objetos o herramientas.
Por otro lado, algunas secuencias de ADN desempeñan un papel estructural en los cromosomas: los telómeros y centrómeros contienen pocos o ningún gen codificante de proteínas, pero son importantes para estabilizar la estructura de los cromosomas. Algunos genes no codifican proteínas, pero sí se transcriben en ARN: ARN ribosómico, ARN de transferencia y ARN de interferencia (ARNi, que son ARN que bloquean la expresión de genes específicos). La estructura de intrones y exones de algunos genes (como los de inmunoglobulinas y protocadherinas) son importantes por permitir los cortes y empalmes alternativos del pre-ARN mensajero que hacen posible la síntesis de diferentes proteínas a partir de un mismo gen (sin esta capacidad no existiría el sistema inmune, por ejemplo). Algunas secuencias de ADN no codificante representan pseudogenes que tienen valor evolutivo, ya que permiten la creación de nuevos genes con nuevas funciones. Otros ADN no codificantes proceden de la duplicación de pequeñas regiones del ADN; esto tiene mucha utilidad, ya que el rastreo de estas secuencias repetitivas permite estudios de filogenia.
En un gen, la secuencia de nucleótidos a lo largo de una hebra de ADN se transcribe a un ARN mensajero (ARNm) y esta secuencia a su vez se traduce a una proteína que un organismo es capaz de sintetizar o "expresar" en uno o varios momentos de su vida, usando la información de dicha secuencia.
La relación entre la secuencia de nucleótidos y la secuencia de aminoácidos de la proteína viene determinada por el código genético, que se utiliza durante el proceso de traducción o síntesis de proteínas. La unidad codificadora del código genético es un grupo de tres nucleótidos (triplete), representado por las tres letras iniciales de las bases nitrogenadas (por ej., ACT, CAG, TTT). Los tripletes del ADN se transcriben en sus bases complementarias en el ARN mensajero, y en este caso los tripletes se denominan codones (para el ejemplo anterior, UGA, GUC, AAA). En el ribosoma cada codón del ARN mensajero interacciona con una molécula de ARN de transferencia (ARNt o tRNA) que contenga el triplete complementario, denominado anticodón. Cada ARNt porta el aminoácido correspondiente al codón de acuerdo con el código genético, de modo que el ribosoma va uniendo los aminoácidos para formar una nueva proteína de acuerdo con las "instrucciones" de la secuencia del ARNm. Existen 64 codones posibles, por lo cual corresponde más de uno para cada aminoácido (por esta duplicidad de codones se dice que el código genético es un código degenerado: no es unívoco); algunos codones indican la terminación de la síntesis, el fin de la secuencia codificante; estos codones de terminación o codones de parada son UAA, UGA y UAG (en inglés, nonsense codons o stop codons).
La replicación del ADN es el proceso por el cual se obtienen copias o réplicas idénticas de una molécula de ADN. La replicación es fundamental para la transferencia de la información genética de una generación a la siguiente y, por ende, es la base de la herencia. El mecanismo consiste esencialmente en la separación de las dos hebras de la doble hélice, las cuales sirven de molde para la posterior síntesis de cadenas complementarias a cada una de ellas, que llevará por nombre ARNm. El resultado final son dos moléculas idénticas a la original. Este tipo de replicación se denomina semiconservativa debido a que cada una de las dos moléculas resultantes de la duplicación presenta una cadena procedente de la molécula "madre" y otra recién sintetizada.
En un principio, se propusieron tres hipótesis:
Todas las funciones del ADN dependen de sus interacciones con proteínas. Estas interacciones pueden ser inespecíficas, o bien la proteína puede unirse de forma específica a una única secuencia de ADN. También pueden unirse enzimas, entre las cuales son particularmente importantes las polimerasas, que copian las secuencia de bases del ADN durante la transcripción y la replicación.
Las proteínas estructurales que se unen al ADN son ejemplos bien conocidos de interacciones inespecíficas ADN-proteínas. En los cromosomas, el ADN se encuentra formando complejos con proteínas estructurales. Estas proteínas organizan el ADN en una estructura compacta denominada cromatina. En eucariotas esta estructura implica la unión del ADN a un complejo formado por pequeñas proteínas básicas denominadas histonas, mientras que en procariotas están involucradas una gran variedad de proteínas. Las histonas forman un complejo de forma cilíndrica denominado nucleosoma, en torno al cual se enrollan casi dos vueltas de ADN de doble hélice. Estas interacciones inespecíficas quedan determinadas por la existencia de residuos básicos en las histonas, que forman enlaces iónicos con el esqueleto de azúcar-fosfato del ADN y, por tanto, son en gran parte independientes de la secuencia de bases. Estos aminoácidos básicos experimentan modificaciones químicas de metilación, fosforilación y acetilación, que alteran la fuerza de la interacción entre el ADN y las histonas, haciendo al ADN más o menos accesible a los factores de transcripción y por tanto modificando la tasa de transcripción.
Otras proteínas que se unen a ADN de manera inespecífica en la cromatina incluyen las proteínas del grupo de alta movilidad (HMG, High Mobility Group) que se unen a ADN plegado o distorsionado. Estas proteínas son importantes durante el plegamiento de los nucleosomas, organizándolos en estructuras más complejas para constituir los cromosomas durante el proceso de condensación cromosómica. Se ha propuesto que en este proceso también intervendrían otras proteínas, formando una especie de "andamio" sobre el cual se organiza la cromatina; los principales componentes de esta estructura serían la enzima topoisomerasa II α (topoIIalpha) y la condensina 13S. Sin embargo, el papel estructural de la topoIIalpha en la organización de los cromosomas aún es discutido, ya que otros grupos argumentan que esta enzima se intercambia rápidamente tanto en los brazos cromosómicos como en los cinetocoros durante la mitosis.
Un grupo bien definido de proteínas que unen ADN es el conformado por las proteínas que se unen específicamente a ADN monocatenario o ADN de hebra sencilla (ssDNA). En humanos, la proteína A de replicación es la mejor conocida de su familia y actúa en procesos en los que la doble hélice se separa, como la replicación del ADN, la recombinación o la reparación del ADN. Estas proteínas parecen estabilizar el ADN monocatenario, protegiéndolo para evitar que forme estructuras de tallo-lazo (stem-loop) o que sea degradado por nucleasas.
Sin embargo, otras proteínas han evolucionado para unirse específicamente a secuencias particulares de ADN. La especificidad de la interacción de las proteínas con el ADN procede de los múltiples contactos con las bases de ADN, lo que les permite "leer" la secuencia del ADN. La mayoría de esas interacciones con las bases ocurre en la hendidura mayor, donde las bases son más accesibles.
Las proteínas específicas estudiadas con mayor detalle son las encargadas de regular la transcripción, denominadas por ello factores de transcripción. Cada factor de transcripción se une a una secuencia concreta de ADN y activa o inhibe la transcripción de los genes que presentan estas secuencias próximas a sus promotores. Los factores de transcripción pueden efectuar esto de dos formas:
Como los ADN diana pueden encontrarse por todo el genoma del organismo, los cambios en la actividad de un tipo de factor de transcripción pueden afectar a miles de genes. En consecuencia, estas proteínas son frecuentemente las dianas de los procesos de transducción de señales que controlan las respuestas a cambios ambientales o diferenciación y desarrollo celular.
Las nucleasas son enzimas que cortan las hebras de ADN mediante la catálisis de la hidrólisis de los enlaces fosfodiéster. Las nucleasas que hidrolizan nucleótidos a partir de los extremos de las hebras de ADN se denominan exonucleasas, mientras que las endonucleasas cortan en el interior de las hebras. Las nucleasas que se utilizan con mayor frecuencia en biología molecular son las enzimas de restricción, endonucleasas que cortan el ADN por determinadas secuencias específicas. Por ejemplo, la enzima EcoRV, que se muestra a la izquierda, reconoce la secuencia de 6 bases 5′-GAT|ATC-3′, y hace un corte en ambas hebras en la línea vertical indicada, generando dos moléculas de ADN con los extremos romos. Otras enzimas de restricción generan, sin embargo, extremos cohesivos, ya que cortan de forma diferente las dos hebras de ADN. En la naturaleza, estas enzimas protegen a las bacterias contra las infecciones de fagos, al digerir el ADN de dicho fago cuando entra a través de la pared bacteriana, actuando como un mecanismo de defensa. En biotecnología, estas nucleasas específicas de la secuencias de ADN se utilizan en ingeniería genética para clonar fragmentos de ADN y en la técnica de huella genética.
Las enzimas denominadas ADN ligasas pueden reunir hebras de ADN cortadas o rotas. Las ligasas son particularmente importantes en la replicación de la hebra que sufre replicación discontinua en el ADN, ya que unen los fragmentos cortos de ADN generados en la horquilla de replicación para formar una copia completa del molde de ADN. También se utilizan en la reparación del ADN y en procesos de recombinación genética.
Las topoisomerasas son enzimas que poseen a la vez actividad nucleasa y ligasa. Estas proteínas varían la cantidad de ADN superenrollado. Algunas de estas enzimas funcionan cortando la hélice de ADN y permitiendo que una sección rote, de manera que reducen el grado de superenrollamiento. Una vez hecho esto, la enzima vuelve a unir los fragmentos de ADN. Otros tipos de enzimas son capaces de cortar una hélice de ADN y luego pasar la segunda hebra de ADN a través de la rotura, antes de reunir las hélices. Las topoisomerasas son necesarias para muchos procesos en los que interviene el ADN, como la replicación del ADN y la transcripción.
Las helicasas son unas proteínas que pertenecen al grupo de los motores moleculares. Utilizan energía química almacenada en los nucleósidos trifosfatos, fundamentalmente ATP, para romper puentes de hidrógeno entre bases y separar la doble hélice de ADN en hebras simples. Estas enzimas son esenciales para la mayoría de los procesos en los que las enzimas necesitan acceder a las bases del ADN.
Las polimerasas son enzimas que sintetizan cadenas de nucleótidos a partir de nucleósidos trifosfatos. La secuencia de sus productos son copias de cadenas de polinucleótidos existentes, que se denominan moldes. Estas enzimas funcionan añadiendo nucleótidos al grupo hidroxilo en 3' del nucleótido previo en una hebra de ADN. En consecuencia, todas las polimerasas funcionan en dirección 5′ → 3′. En los sitios activos de estas enzimas, el nucleósido trifosfato que se incorpora aparea su base con la correspondiente en el molde: esto permite que la polimerasa sintentice de forma precisa la hebra complementaria al molde.
Las polimerasas se clasifican de acuerdo al tipo de molde que utilizan:
Una hélice de ADN normalmente no interacciona con otros segmentos de ADN, y en las células humanas los diferentes cromosomas incluso ocupan áreas separadas en el núcleo celular denominadas “territorios cromosómicos”. La separación física de los diferentes cromosomas es importante para que el ADN mantenga su capacidad de funcionar como un almacén estable de información. Uno de los pocos momentos en los que los cromosomas interaccionan es durante el sobrecruzamiento cromosómico (chromosomal crossover), durante el cual se recombinan. El sobrecruzamiento cromosómico ocurre cuando dos hélices de ADN se rompen, se intercambian y se unen de nuevo.
La recombinación permite a los cromosomas intercambiar información genética y produce nuevas combinaciones de genes, lo que aumenta la eficiencia de la selección natural y puede ser importante en la evolución rápida de nuevas proteínas. Durante la profase I de la meiosis, una vez que los cromosomas homólogos están perfectamente apareados formando estructuras llamadas bivalentes, se produce el fenómeno de sobrecruzamiento o entrecruzamiento (crossing-over), en el cual las cromátidas homólogas no hermanas (procedentes del padre y de la madre) intercambian material genético. La recombinación genética resultante hace aumentar en gran medida la variación genética entre la descendencia de progenitores que se reproducen por vía sexual. La recombinación genética también puede estar implicada en la reparación del ADN, en particular en la respuesta celular a las roturas de doble hebra (double-strand breaks).
La forma más frecuente de sobrecruzamiento cromosómico es la recombinación homóloga, en la que los dos cromosomas implicados comparten secuencias muy similares. La recombinación no-homóloga puede ser dañina para las células, ya que puede producir translocaciones cromosómicas y anomalías genéticas. La reacción de recombinación está catalizada por enzimas conocidas como recombinasas, tales como RAD51. El primer paso en el proceso de recombinación es una rotura de doble hebra, causada bien por una endonucleasa o por daño en el ADN. Posteriormente, una serie de pasos catalizados en parte por la recombinasa, conducen a la unión de las dos hélices formando al menos una unión de Holliday, en la que un segmento de una hebra simple es anillado con la hebra complementaria en la otra hélice. La unión de Holliday es una estructura de unión tetrahédrica que puede moverse a lo largo del par de cromosomas, intercambiando una hebra por otra. La reacción de recombinación se detiene por el corte de la unión y la reunión de los segmentos de ADN liberados.
El ADN contiene la información genética que permite a la mayoría de los organismos vivientes funcionar, crecer y reproducirse. Sin embargo, no está claro durante cuánto tiempo ha ejercido esta función en los ~3000 millones de años de la historia de la vida, ya que se ha propuesto que las formas de vida más tempranas podrían haber utilizado ARN como material genético. El ARN podría haber funcionado como la parte central de un metabolismo primigenio, ya que puede transmitir información genética y simultáneamente actuar como catalizador formando parte de las ribozimas. Este antiguo Mundo de ARN donde los ácidos nucleicos funcionarían como catalizadores y como almacenes de información genética podría haber influido en la evolución del código genético actual, basado en cuatro nucleótidos. Esto se debería a que el número de bases únicas en un organismo es un compromiso entre un número pequeño de bases (lo que aumentaría la precisión de la replicación) y un número grande de bases (que a su vez aumentaría la eficiencia catalítica de las ribozimas).
Desafortunadamente, no se cuenta con evidencia directa de los sistemas genéticos ancestrales porque la recuperación del ADN a partir de la mayor parte de los fósiles es imposible. Esto se debe a que el ADN es capaz de sobrevivir en el medio ambiente durante menos de un millón de años, y luego empieza a degradarse lentamente en fragmentos de menor tamaño en solución. Algunas investigaciones pretenden que se ha obtenido ADN más antiguo, por ejemplo un informe sobre el aislamiento de una bacteria viable a partir de un cristal salino de 250 millones de años de antigüedad, pero estos datos son controvertidos.
Sin embargo, pueden utilizarse herramientas de evolución molecular para inferir los genomas de organismos ancestrales a partir de organismos contemporáneos. En muchos casos, estas inferencias son suficientemente fiables, de manera que una biomolécula codificada en un genoma ancestral puede resucitarse en el laboratorio para ser estudiada hoy. Una vez que la biomolécula ancestral se ha resucitado, sus propiedades pueden ofrecer inferencias sobre ambientes y estilos de vida primigenios. Este proceso se relaciona con el campo emergente de la paleogenética experimental.
A pesar de todo, el proceso de trabajo hacia atrás desde el presente tiene limitaciones inherentes, razón por la cual otros investigadores tratan de elucidar el mecanismo evolutivo trabajando desde el origen de la Tierra en adelante. Dada suficiente información sobre la química en el cosmos, la manera en la que las sustancias cósmicas podrían haberse depositado en la Tierra, y las transformaciones que podrían haber tenido lugar en la superficie terrestre primigenia, tal vez podríamos ser capaces de aprender sobre los orígenes para desarrollar modelos de evolución ulterior de la información genética (véase también el artículo sobre el origen de la vida).
El conocimiento de la estructura del ADN ha permitido el desarrollo de multitud de herramientas tecnológicas que explotan sus propiedades fisicoquímicas para analizar su implicación en problemas concretos: por ejemplo, desde análisis filogeńeticos para detectar similitudes entre diferentes taxones, a la caracterización de la variabilidad individual de un paciente en su respuesta a un determinado fármaco, pasando por un enfoque global, a nivel genómico, de cualquier característica específica en un grupo de individuos de interés.
Podemos clasificar las metodologías de análisis del ADN en aquellas que buscan su multiplicación, ya in vivo, como la reacción en cadena de la polimerasa (PCR), ya in vitro, como la clonación, y aquellas que explotan las propiedades específicas de elementos concretos, o de genomas adecuadamente clonados. Es el caso de la secuenciación de ADN y de la hibridación con sondas específicas (Southern blot y chips de ADN).
La tecnología del ADN recombinante, piedra angular de la ingeniería genética, permite propagar grandes cantidades de un fragmento de ADN de interés, el cual se dice que ha sido clonado. Para ello, debe introducirse dicho fragmento en otro elemento de ADN, generalmente un plásmido, que posee en su secuencia los elementos necesarios para que la maquinaria celular de un hospedador, normalmente Escherichia coli, lo replique. De este modo, una vez transformada la cepa bacteriana, el fragmento de ADN clonado se reproduce cada vez que aquella se divide.
Para clonar la secuencia de ADN de interés, se emplean enzimas como herramientas de corte y empalme del fragmento y del vector (el plásmido). Dichas enzimas corresponden a dos grupos: en primer lugar, las enzimas de restricción, que poseen la capacidad de reconocer y cortar secuencias específicas; en segundo lugar, la ADN ligasa, que establece un enlace covalente entre extremos de ADN compatibles (ver sección Nucleasas y ligasas).
La secuenciación del ADN consiste en dilucidar el orden de los nucleótidos de un polímero de ADN de cualquier longitud, si bien suele dirigirse hacia la determinación de genomas completos, debido a que las técnicas actuales permiten realizar esta secuenciación a gran velocidad, lo cual ha sido de gran importancia para proyectos de secuenciación a gran escala como el Proyecto Genoma Humano. Otros proyectos relacionados, en ocasiones fruto de la colaboración de científicos a escala mundial, han establecido la secuencia completa del ADN de muchos genomas de animales, plantas y microorganismos.
El método de secuenciación de Sanger ha sido el más empleado durante el siglo XX. Se basa en la síntesis de ADN en presencia de didesoxinucleósidos, compuestos que, a diferencia de los desoxinucleósidos normales (dNTPs), carecen de un grupo hidroxilo en su extremo 3'. Aunque los didesoxinucleótidos trifosfatados (ddNTPs) pueden incorporarse a la cadena en síntesis, la carencia de un extremo 3'-OH imposibilita la generación de un nuevo enlace fosfodiéster con el nucleósido siguiente; por tanto, provocan la terminación de la síntesis. Por esta razón, el método de secuenciación también se denomina «de terminación de cadena». La reacción se realiza usualmente preparando un tubo con el ADN molde, la polimerasa, un cebador, dNTPs convencionales y una pequeña cantidad de ddNTPs marcados fluorescentemente en su base nitrogenada. De este modo, el ddTTP puede ir marcado en azul, el ddATP en rojo, etc. Durante la polimerización, se van truncando las cadenas crecientes, al azar, en distintas posiciones. Por tanto, se produce una serie de productos de distinto tamaño, coincidiendo la posición de la terminación debido a la incorporación del ddNTP correspondiente. Una vez terminada la reacción, es posible correr la mezcla en una electroforesis capilar (que resuelve todos los fragmentos según su longitud) en la cual se lee la fluorescencia para cada posición del polímero. En nuestro ejemplo, la lectura azul-rojo-azul-azul se traduciría como TATT.
La reacción en cadena de la polimerasa, habitualmente conocida como PCR por sus siglas en inglés, es una técnica de biología molecular descrita en 1986 por Kary Mullis, cuyo objetivo es obtener un gran número de copias de un fragmento de ADN dado, partiendo de una escasa cantidad de aquel. Para ello, se emplea una ADN polimerasa termoestable que, en presencia de una mezcla de los cuatro desoxinucleótidos, un tampón de la fuerza iónica adecuada y los cationes precisos para la actividad de la enzima, dos oligonucleótidos (denominados cebadores) complementarios a parte de la secuencia (situados a distancia suficiente y en sentido antiparalelo) y bajo unas condiciones de temperatura adecuadas, moduladas por un aparato denominado termociclador, genera exponencialmente nuevos fragmentos de ADN semejantes al original y acotados por los dos cebadores.
La PCR puede efectuarse como una técnica de punto final, esto es, como una herramienta de generación del ADN deseado, o como un método continuo, en el que se evalúe dicha polimerización a tiempo real. Esta última variante es común en la PCR cuantitativa.
El método de «hibridación Southern» o Southern blot (el nombre original en el idioma inglés) permite la detección de una secuencia de ADN en una muestra compleja o no del ácido nucleico. Para ello, combina una separación mediante masa y carga (efectuada mediante una electroforesis en gel) con una hibridación con una sonda de ácido nucleico marcada de algún modo (ya sea con radiactividad o con un compuesto químico) que, tras varias reacciones, dé lugar a la aparición de una señal de color o fluorescencia. Dicha hibridación se realiza tras la transferencia del ADN separado mediante la electroforesis a una membrana de filtro. Una técnica semejante, pero en la cual no se produce la mencionada separación electroforética se denomina dot blot.
El método recibe su nombre en honor a su inventor, el biólogo inglés Edwin Southern. Por analogía al método Southern, se han desarrollado técnicas semejantes que permiten la detección de secuencias dadas de ARN (método Northern, que emplea sondas de ARN o ADN marcadas) o de proteínas específicas (técnica Western, basada en el uso de anticuerpos).
Los chips de ADN son colecciones de oligonucleótidos de ADN complementario dispuestos en hileras fijadas sobre un soporte, frecuentemente de cristal. Se utilizan para el estudio de mutaciones de genes conocidos o para monitorizar la expresión génica de una preparación de ARN.
La investigación sobre el ADN tiene un impacto significativo, especialmente en el ámbito de la medicina, pero también en agricultura y ganadería (donde los objetivos son los mismos que con las técnicas tradicionales que el hombre lleva utilizando desde hace milenios —la domesticación, la selección y los cruces dirigidos— para obtener variedades de animales y plantas más productivos). La moderna biología y bioquímica hacen uso intensivo de la tecnología del ADN recombinante, introduciendo genes de interés en organismos, con el objetivo de expresar una proteína recombinante concreta, que puede ser:
Los médicos forenses pueden utilizar el ADN presente en la sangre, el semen, la piel, la saliva o el pelo en la escena de un crimen, para identificar al responsable. Esta técnica se denomina huella genética, o también "perfil de ADN". Al realizar la huella genética, se compara la longitud de secciones altamente variables de ADN repetitivo, como los microsatélites, entre personas diferentes. Este método es frecuentemente muy fiable para identificar a un criminal. Sin embargo, la identificación puede complicarse si la escena está contaminada con ADN de personas diferentes. La técnica de la huella genética fue desarrollada en 1984 por el genetista británico sir Alec Jeffreys, y fue utilizada por primera vez en medicina forense para condenar a Colin Pitchfork por los asesinatos de Narborough en 1983 y de Enderby en 1986. Se puede requerir a las personas acusadas de ciertos tipos de crímenes que proporcionen una muestra de ADN para introducirlos en una base de datos. Esto ha facilitado la labor de los investigadores en la resolución de casos antiguos, donde solo se obtuvo una muestra de ADN de la escena del crimen, en algunos casos permitiendo exonerar a un convicto. La huella genética también puede utilizarse para identificar víctimas de accidentes en masa, o para realizar pruebas de consanguinidad (prueba de paternidad).
La bioinformática implica la manipulación, búsqueda y extracción de información de los datos de la secuencia del ADN. El desarrollo de las técnicas para almacenar y buscar secuencias de ADN ha generado avances en el desarrollo de software de los ordenadores, para muchas aplicaciones, especialmente algoritmos de búsqueda de frases, aprendizaje automático y teorías de bases de datos. La búsqueda de frases o algoritmos de coincidencias, que buscan la ocurrencia de una secuencia de letras dentro de una secuencia de letras mayor, se desarrolló para buscar secuencias específicas de nucleótidos. En otras aplicaciones como editores de textos, incluso algoritmos simples pueden funcionar, pero las secuencias de ADN pueden generar que estos algoritmos presenten un comportamiento de casi-el-peor-caso, debido al bajo número de caracteres. El problema relacionado del alineamiento de secuencias persigue identificar secuencias homólogas y localizar mutaciones específicas que las diferencian. Estas técnicas, fundamentalmente el alineamiento múltiple de secuencias, se utilizan al estudiar las relaciones filogenéticas y la función de las proteínas. Las colecciones de datos que representan secuencias de ADN del tamaño de un genoma, tales como las producidas por el Proyecto Genoma Humano, son difíciles de usar sin anotaciones, que marcan la localización de los genes y los elementos reguladores en cada cromosoma. Las regiones de ADN que tienen patrones asociados con genes que codifican proteínas —o ARN— pueden identificarse por algoritmos de localización de genes, lo que permite a los investigadores predecir la presencia de productos génicos específicos en un organismo incluso antes de que haya sido aislado experimentalmente.
La nanotecnología de ADN utiliza las propiedades únicas de reconocimiento molecular del ADN y otros ácidos nucleicos para crear complejos ramificados auto-ensamblados con propiedades útiles. En este caso, el ADN se utiliza como un material estructural, más que como un portador de información biológica. Esto ha conducido a la creación de láminas periódicas de dos dimensiones (ambas basadas en azulejos, así como usando el método de ADN origami), además de estructuras en tres dimensiones con forma de poliedros.
A lo largo del tiempo, el ADN almacena mutaciones que se heredan y, por tanto, contiene información histórica, de manera que comparando secuencias de ADN, los genetistas pueden inferir la historia evolutiva de los organismos, su filogenia. La investigación filogenética es una herramienta fundamental en biología evolutiva. Si se comparan las secuencias de ADN dentro de una especie, los genetistas de poblaciones pueden conocer la historia de poblaciones particulares. Esto se puede utilizar en una amplia variedad de estudios, desde ecología hasta antropología, como ilustra el análisis de ADN llevado a cabo para identificar las Diez Tribus Perdidas de Israel. Por otro lado, el ADN también se utiliza para estudiar relaciones familiares recientes.
Igualmente en paleontología (en la paleogenética) el ADN en algunos casos también se puede utilizar para estudiar a especies extintas (ADN fósil).
Escribe un comentario o lo que quieras sobre Ácido desoxirribonucleico (directo, no tienes que registrarte)
Comentarios
(de más nuevos a más antiguos)
Aún no hay comentarios, ¡deja el primero!